
-
首页
-
DIA年会
-
专题会议
-
线下培训
-
线上课程
-
DIA介绍
- DIA历史
- DIA 国际版
- DIA全球董事会成员
- DIA中国顾问委员会成员
- DIA全球会士
-
DIA社区
-
DIA全球会员
-
学术电子词典
- 统计、药物警戒电子词典
- Global Forum中文版
- 蓝皮书
-
首页
-
DIA年会
-
专题会议
-
线下培训
-
线上课程
-
DIA介绍
- DIA历史
- DIA 国际版
- DIA全球董事会成员
- DIA中国顾问委员会成员
- DIA全球会士
-
DIA社区
-
DIA全球会员
-
学术电子词典
- 统计、药物警戒电子词典
- Global Forum中文版
- 蓝皮书
前言/
8月10日中午 DIA中国药品法规事务社区(RAC)组织了“欧美法规热点进行时”第五期直播,主题为《The Basics of labeling in the US》,邀请的讲者是来自BRIM Biotechnology, Inc. (全福生物科技)的法规专家Su-Yueh Lin老师。Su老师在药品说明书领域的经验非常丰富,利用简短的一小时把US labeling大框架进行了梳理和分享,让线上观众有种茅塞顿开的感觉。感谢iReg将直播精华做了总结和梳理,形成此听课笔记做分享。文末附线上问答集锦。
“欧美法规热点进行时”系列活动预告
|
April |
|
|
May |
|
|
June |
|
|
July |
|
|
August |
|
|
September |
|

加入DIA全球会员,
获取直播回放观看权益
扫描左侧二维码
首先有必要说明下label和labeling的区别:label指标签,例如贴在Vial上的标签。labeling包括所有和药品说明书、包装相关的文件。US labeling包括Prescribing Information (PI)、Medication Guide (MG)、Patient Package Insert (PPI)、Instructions For Use documents (IFU)等;同时,与药品说明书、内外标签相关的文件,都可以称之为labeling。Labeling 包含了label,比label的概念要广。
1
美国处方药labeling有关文件简介
Prescribing Information(PI)
主要是给医疗工作者看的,含有最核心的数据,所有处方药都有,格式也是固定的,是不可少的文件。
Medication Guide(MG)
主要是给患者看的,不是必须的。当药品有一定的风险,例如 prescribing information中有warning box,有预防严重不良反应的重要信息,FDA 可能要求企业提供MG。如Su老师所讲,大部分MG是REMS(Risk Evaluation and Mitigation Strategy)的一部分,是为了控制药品使用时的风险。MG最后成文需要FDA的批准。MG的格式,可参考FDA法规 21 CRF 208。
Patient Package Insert(PPI)
主要给患者看的,除了口服避孕药和激素类药外(这两类药必须要有PPI),不是必须的。
Instructions for Use documents (IFU)
特殊剂型,例如预充针(pre-filled syringe)等需要有这个文件,FDA可能要求做User Testing。
Consumer Medication Information
这个FDA没有要求,一般是第三方准备的,例如药师来准备。
Su老师还提到Patient Medication Information。这个是FDA提出把MG和PPI合并在一起的一个计划,但现在还处于pending之中。
2
Structured Product Labeling (SPL) 简介
SPL是一个XML文件。SPL内容除了包含US PI、MG、PPI、IFU这些文件外,还包含了标签和外包装以及data element。之前我们说过的labeling文件,都包含在里面。
SPL是XML格式的,FDA网站上有生成SPL文件的工具和指南。如果大家看过FDA的NDA批件,上面总有一段这样的话:药品批准后的14天内,必须提交SPL。
NDA/BLA中,需要提交三种格式的labeling文件:word版、PDF版以及SPL版。
Su老师解释了这三种格式:word文件是提供给审评员以方便修改的;PDF是为了固定word内容以方便查看(word在不同电脑上打开看到的格式可能会变动,但PDF不受电脑影响);SPL是为了证明企业有生成这个XML文件的能力。此外,药品生产者要求使用SPL,去Electronic Drug Registration and Listing System (eDRLS)登记信息。可以说,SPL也是FDA Drug listing的一部分。
3
查询Labeling的三个数据库
Su老师介绍了三个数据库:Drugs@FDA、DailyMed和 FDALabel。三个数据库的不同点,Su老师做了如下对比。三个数据库各有特色和差异,可根据自己的需求选择数据库进行查询。
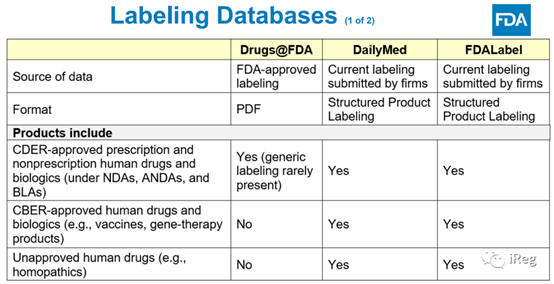
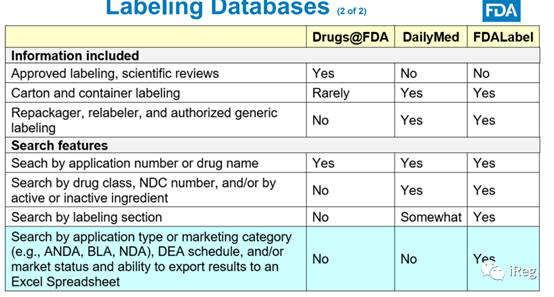
Su老师增加了两个额外的知识点:
⑴. FDA-Approved Labeling 和 Current Labeling的定义:FDA-Approved Labeling是指被FDA批准并在Drugs@FDA公开的Labeling,而Current Labeling不等于FDA-Approved Labeling,有可能在FDA-Approved Labeling后,企业又做了一些小变更。
⑵. CBE-30(Changes Being Effected in 30 Days)类型只适用于CMC变更,CBE-0(Changes Being Effected 0,也称为CBE)是对labeling的变更。除了CBE-0,labeling上市后变更,还可以走年报和PAS(Prior Approval Supplement)途径。
4
FDA Labeling Policy Team
Labeling Policy Team (LPT) 隶属于FDA 新药办公室,负责新药审评中有关labeling的各项工作。Labeling中的内容涉及到各个学科,FDA内部需要进行跨部门合作,LPT负责内部协调。
Labeling的形式审核结果会发布在NDA/BLA 审评的day -74 letter里,申请者可按要求重新提交符合要求的labeling文件。在NDA/BLA批准前4-5周,FDA和申请者会就labeling文件内容进行反复的negotiation,最终达到FDA和申请者都同意的版本。
5
Labeling的批准前\后事项
NDA/BLA 批准前:Investigator’s Brochure (IB)可以看成是USPI的前身。Target Product Profile (TPP) 中也包含了USPI的关键信息,但TPP指南已被FDA撤回。
药品上市后:根据21 CFR 201.56(a)(2),如出现药品新的信息,导致labeling上的内容不准确、错误或是有误导性,都应该进行labeling变更。
6
和国内的异同点
1. 国内没有SPL的要求。
2. 没有各种各样的labeling,只有药品说明书、标签和外包装。
3. 说明书查询,多是民间的数据库,没有大而全的官方说明书查询数据库。例如江苏省的上市药品,可到江苏省药监督局查询。此外,一些行业内的数据库(如药智网)也可查询,可参考见之前iReg发表的文章:世界各国药品说明书查询——中国篇之一“网”打尽
4. 说明书核准是一样的。国内新药批准前,CDE也需要和申请者进行多轮的说明书核准(Labeling negotiation)。
作者致谢
之前也研究过一些US labeling的法规,零零星星的,拼凑不起来。经过Su老师的梳理,恍然大悟的感觉。最后感谢DIA药品法规事务社区提供这么好的学习资源,收获颇丰。
以下是来自直播间听众的提问和讲者的解答:
Q
请问FDA修订有关药品说明书指南的流程是怎样的呢?
A
FDA通常会发布各种指南制定计划,告诉大家涉及哪些类别。先从概念到制定Draft版本,经过一段时间的测试来确定是否需要调整,最后再确定Final版本。这个指南不仅是给申办方提供详细的信息,帮助大家弄清楚如何解决具体问题,同时也是给FDA的审评专家做参考,规范审评标准。
Q
FDA在审评说明书的时,会同时审评SPL文件以及递交的其他格式的文件么?
A
FDA要求递交申请时需要提交SPL文件(XML),PDF和word文件。但是,实际我们在和FDA沟通时往来的回复函通常使用的是Word版本。我觉得SPL文件最要重要作用不是在递交Submission阶段,而是在获得FDA批准之后。FDA批件中有明确规定要在要批获批后14天内提交SPL。
Q
Medication Guide (MG) 不是必须递交的,那么请问如何确定什么情况下需要准备MG?是否会在pre-NDA meeting中和FDA 商量确定?
A
MG其实是数据驱动的,通常是有涉及特定的种族人群、有非常特定的风险或黑框警告时才会准备MG。除这些情况外,准备PPI就可以了,PPI中包含对一般适应症和风险的描述。所以,如果对于没有特别风险的新药,通常不会主动要求去递交MG。
Q
请问Su老师,Medication Guide是什么产品需要呢?
A
Medication Guide 只针对某些特定的药物或特定类别的药物,如果FDA 认为此申请的药物有以下的情况与风险,会要求Medication Guide:
1. 必需传答某些信息来避免严重的副作用
2. 病人需被告知严重的副作用来做判断
3. 病人必需尊循药物的使用方法才能确保药物的疗效
Q
临床样品label有没有格式和内容的要求?临床样品bottle label上有效期怎么定?
A
临床样品的label与 prescription drug labeling 是不同的,所以我并不熟悉,我查看了一下e-CFR,有下面信息。
临床样品bottle label上有效期应是根据制造厂稳定性研究结果定的储存期限以及药品制造日期来决定。
https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/CFRSearch.cfm?fr=312.6
以下业界的网页也有相关临床样品label的资讯与建议可做参考(2021年6月10日公布),也引用了美国用药安全研究所(Institute for Safe Medication Practices,ISMP)的建议
https://www.luminer.com/articles/regulations-for-clinical-trial-labeling/
Q
请问USPI在得到批准后,多久时间会publish到FDA网站上?
A
● Drugs@FDA:约 2-3 天可看到 FDA 的 Approval Letter和FDA-approved Labeling.
● DailyMed和FDALabel:药厂在收到 FDA Approval Letter 当天起 2 星期 (14 days) 内必需通过E-gateway递交SPL完成 Drug Listing 首次登记或变更。在 SPL 成功受理后 (建议每天查看 E-Gateway 有沒有收到 error message),约 2-3 天可在DailyMed 上 查找到"Current Labeling" 及其更新日期,若一星期后还没更新,建议尽快联系 eDRLS@fda.hhs.gov。
编辑:iReg
▎
关于DIA