
DIA年会洞察|紧跟前沿精髓,推动跨越发展——转化医学社区专场(伴随诊断法规篇)
2022 DIA年会
在刚刚结束的2022年DIA中国年会上,转化医学专场在社区志愿者的共同努力下,顺利开展。本次社区的专题会议内容分成两个模块进行,来自学术界、工业界及科研界的各位专家同道们就转化医学在相关领域的前沿发展和行业发展阶段的影响模式,进行了深入的讨论。虽然整个行业都受到疫情影响,线下会议举办受到条件局限,但是,我们对转化医学的热情互动并未因此而减少,整个大会采用全程线上直播及录播的方式进行,邀请到行业的科学家及领域中的相关企业参与讨论。
此次转化医学年会环节的主题分两个部分:
|
转化医学之法规篇章 |
伴随诊断新法规探索 |
|
转化医学之合作模式篇章 |
产、科、研三方合作模式探讨 |
在转化医学法规篇章中,大会有幸邀请到几位行业资深专家做了相关主题分享:
首先开场演讲的是来自国家医疗器械技术评审中心的临床与统计二部的方丽老师。方老师的演讲主题是:抗肿瘤药物的伴随诊断试剂相关指导原则的介绍。
最近几年肿瘤药物研发过程中的伴随诊断的研发一直是行业热点,随着中国相关法规的不断更新出台,为行业的相关企业的执行过程给出了及时、细致的指导原则,此次大会荣幸的邀请到方丽老师就这些新的法规做出了精彩的解读。
与抗肿瘤药物相关的伴随诊断试剂是对采集自肿瘤患者的样本进行检测,其检测结果可以为患者使用抗肿瘤药物的安全性和有效性提供重要的信息。主要有三大作用,即:确定最有可能从药物中受益的患者;确定该药物相关严重不良反应风险较大的患者;确定已经过充分研究具备安全性和有效性的人群亚组。在国内已经批准的伴随诊断试剂较多,从方法学上可以分为:免疫组化、荧光原位杂交;高通量测序等多种方法学均有涵盖,所涉及相关的抗肿瘤药物也涵盖了抗体类药物和小分子药物,为临床的诊疗需求提供了一定程度上的支持。

伴随诊断试剂的开发模式,可以分为两大类:原研伴随诊断试剂与抗肿瘤药物共同开发,及抗肿瘤药物已上市的非原研伴随诊断试剂的开发。方丽老师就这两种不同类型的开发模式,如何进行验证与研究进行了介绍。
与抗肿瘤药物同步研发的临床试验设计有三种类型可以选择。首先是已设计定型的伴随诊断试剂,直接参与到药物的临床研究过程,也就是说在药物的开发过程中同步进行伴随诊断试剂的开发,在药物非临床及临床研究过程中,通过与伴随诊断试剂共同验证确认,确定药物可能安全有效的治疗人群。第二种方式是在抗肿瘤药物临床实验中采用临床试验分析方法(CTA)来进行病例的生物标志物分析,而相应的伴随诊断试剂的开发过程相较于药物的开发过程是滞后的,采用桥接试验的开发路径。第三种方式是药物开发过程中,选择已经上市的同类产品作为该药物的伴随诊断试剂,参与药物的研发过程中。
在伴随诊断试剂具体产品指导原则的分享解读中,方丽老师就近几年发布的针对具体产品的指导原则进行了详细的拆解,其中2022年发布的《PD-L1检测试剂临床试验-结果重现性研究注册审查指导原则》和2023年即将发布的《基于高通量测序技术的非小细胞肺癌相关基因变异检测试剂临床试验注册审查指导原则》做了针对性的分享。
方丽老师也对比较成熟的伴随诊断产品EGFR(PCR法)类产品如何注册申报进行了详细的解读。EGFR突变基因检测试剂(PCR法),需要明确适应症为非小细胞肺癌,具体的是哪一个TKI抑制剂的药物需要明确说明,对于病例组织样本和外周血样本需要进行分别评价,而且应该纳入所有的声称检测的突变位点,统计分析部分要在两个层面上进行阴阳性符合率的统计,一是总体统计,二是每个突变类别的统计。

《基于高通量测序技术的非小细胞肺癌相关基因变异检测试剂临床试验注册审查指导原则》是第一个基于临床适应症的指导原则,方丽老师也做了分享介绍。其适用的范围是针对采用NGS技术检测NSCLC患者福尔马林固定石蜡包埋组织样本中的基因变异状态的体外诊断试剂,而针对的入组人群需要关注纳入NSCLC不同分期的病例,包括从I期到IV期的患者;同时需要纳入不同的组织分型的患者,比如鳞癌、腺癌、腺鳞癌及大细胞癌等。
第二位做分享的嘉宾来自罗氏诊断(中国)医学法务事务部副总裁尹琦曼女士,尹总拥有二十余年的药品、医疗器械的丰富法规、临床及质量管理的经验。
尹总从中外伴随诊断的注册法规的对比及其中行业所面对的挑战和机遇两个角度进行了分享。尹总的分享从伴随诊断试剂的世界监管地图开篇,全球已经有184个国家和地区拥有自己独立的器械监管体系,在这些国家中只有包括中国在内的少数国家具有单独的独立伴随诊断试剂的相关的法律法规要求,并对于美国及中国的伴随诊断试剂的批准情况进行了系统地比较和分析。
尹总从伴随诊断试剂的法规范畴,定义、标签、分类、临床证据、审批路径这多个综合方面进行了比对介绍。在临床证据方面,中国的相关法规和指导原则并未排除真实世界证据作为临床证据的可能性,只是中国还尚未有通过真实世界的临床证据来支持获批的伴随诊断试剂。而伴随诊断试剂注册的临床证据,在不同的在药品临床和伴随诊断试剂临床中具有不同的目的。药物临床试验需要说明的是伴随诊断试剂与药物疗效的关系问题,相关样本允许转运到同一个临床研究中心实验室完成检测,着眼与伴随诊断试剂的水平与药物最终疗效的关联性的问题。而伴随诊断试剂临床试验则是解决伴随诊断试剂在不同的医疗机构间检测准确性的问题,着眼的是从前处理到结果判断的全过程,医师操作和技术水平对最终结果准确性的影响的验证。这两个研究相互关联,但是互相不可取代。

此外,尹总通过对全球视角下,中西方不同阶段的伴随诊断试剂相关法规的详细比较,阐明了中国的伴随诊断试剂开发中的机遇与挑战并存的现状。药物三期临床试验同时平行开展伴随诊断试剂临床试验,是缩小药品与其伴随诊断试剂审批时间差距的最有效方式。豁免原产国上市要求是缩小进口伴随诊断试剂与药品审批时间差距的长期解决方式。从伴随诊断试剂与药品在中国同步批准的分开审评构想最核心的是“先解决有没有的问题,再解决普及性的问题”。理想的伴随诊断试剂的战略合作伙伴关系是研发早期制药企业与伴随诊断试剂企业进行双向跨界合作。
第三位分享嘉宾是百济神州全球转化研究及转化医学负责人沈志荣博士。沈博士负责百济神州临床前创新项目研发、转化发现研究、临床转化研究及伴随诊断开发。

沈博士结合自己从业十余年的经验,就伴随诊断试剂与创新药物的全球同步开发进行了分享。沈博士通过详细的分析全球同步开发的难点和机会,结合自身经验在药物与伴随诊断的共同开发的实际案例,呈现了他对于伴随诊断全球同步开发的精彩分享。

抗肿瘤新药研发过程中,伴随诊断试剂的必要性已经毋庸置疑。越来越多新模式的中国原创新药类型如靶向抗原的双抗、抗体偶联药物、CAR-T/CAR-NK细胞治疗等蓬勃发展,在这类创新药物的临床开发中伴随诊断变得不可或缺,因此伴随诊断试剂的开发也越来越多的被行业所关注。当前对伴随诊断的全球共同监管的趋势是日益严格,同时在各个国家的不同法规背景下,开发的路径和机遇也具有差异性。对于国内的生物研发企业,在新药研发的早期就开始进行相关国家的伴随诊断开发的提前布局,是具有战略意义的。这些包括了药物伴随诊断试剂的全球各个国家的监管要求细则的解读,临床样本检测的要求,审批路径,适应症限制的趋势及法规等多个详细的内容,都需要研发药企做深入的规划。
沈博士在演讲中,通过具体的实践案例做了生动的分享。药物的临床研发路径常见的是三种类型,即对于富集人群进行单臂临床试验、对于富集人群的随机对照临床试验、对非富集人群的随机对照试验。百济神州的PD-1抗体在早期开发的第一个实体瘤适应症的案例,就是通过PD-L1 的检测针对富集PD-L1阳性的尿路上皮癌的人群,进行单臂临床而获批上市的。对一期临床研究的样本进行回顾性的检测,确定了不同的cut off值。因此在设计单臂二期临床试验时,即采用所选的PD-L1检测和相应的cut off值作为入组标准,最终临床结果显示客观缓解率(ORR)高于历史对照,并获得新药审批上市,且相应的PD-L1检测伴随诊断试剂也获得同步获批。这是首个按照伴随诊断要求,并基于国内自主开展的临床试验获批的伴随诊断,也是百济神州、罗氏诊断和第三方实验室团队三方共同努力的结果。
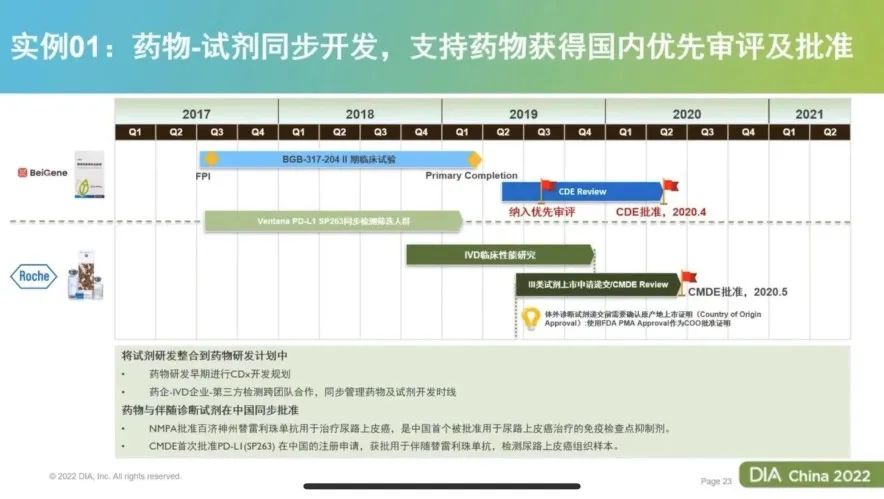
在演讲中,沈博士还介绍了其他多个行业中的经典案例,也为行业的同仁们在新药研发和伴随诊断试剂的共同开发的策略制定和实际应用上提供了非常精彩的参考分享。
本次大会的伴随诊断试剂相关法规专题环节,由罗氏诊断医学法规事务部的蔡晓蓉总监主持。

- 2025-08-15
- 2025-07-11
- 2025-07-04
- 2025-06-27
-
2025-06-20
낑 罕见病基础培训
- 2025-06-13
- 2025-05-20
- 2025-04-18
- 2025-04-17
- 2025-04-11